Myocardial cells have distinctive anatomic and physiologic characteristics. Under a microscope, striations are visible owing to the arrangement of actin and myosin as in skeletal muscle. T tubules increase the contact area of the cell membrane and extracellular fluid space.
At rest, cardiac muscle obtains 99% of its energy from aerobic metabolism. Myocardial cells have numerous mitochondria to support energy production. In addition, high capillary density ensures adequate blood delivery to support aerobic metabolism. Multiple substrates can provide energy for the heart. At rest, 60% of energy is derived from metabolism of fat, 35% from carbohydrates, and 5% from amino acids and ketones.
Myocardial Mechanics
Excitation-contraction coupling ties electrical depolarization to the mechanical shortening of myocardial cells. Myocardial depolarization allows influx of extracellular Ca++ during phase 2 of myocardial action potential. This extracellular Ca++ “triggers” the release of Ca++ from sarcoplasmic reticulum (SR). Over 90% of the Ca++ involved in the contraction comes from the SR. By the end of systole, Ca++ is resequestered in SR, and extracellular Ca++ is removed by an electroneutral 2 Na+/Ca++ exchange.
There is a time delay before Ca++ that is taken up by the SR is available for release by the next depolarization. A premature depolarization has diminished SR Ca++ release and consequently lower than normal force generation. In contrast, enhanced contractile strength characterizes a delayed depolarization because of the additional time for processing of SR Ca++ into the available pool.
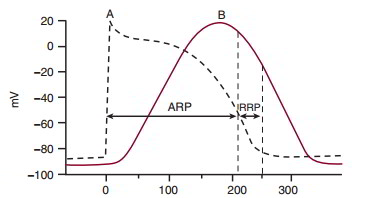
Myocardial cell action potential precedes the contraction. Contraction of the ventricular muscle cell (curve B in above figure) requires the entry of Ca++ during phase 2 of the ventricular muscle action potential (curve A), and relaxation begins during phase 3. During an action potential, the ventricular muscle cell cannot initiate a second action potential, since it is in the absolute refractory period. During the end of the action potential, there is a relative refractory period during which only a greater than normal stimulus can initiate a subsequent action potential. The ventricular muscle cell contraction is completed shortly after this relative refractory period. Consequently, ventricular muscle contraction is a series of twitch contractions, and the myocardium cannot enter tetany.
Contractile activity of the heart can be modeled by the activity of a contractile component and an elastic component in series. The contractile element reflects the action of actin and myosin. A parallel elastic element reflects the tendency of connective tissue to resist stretch, so stretch causes some tension in the muscle even without a contraction. The series elastic element has no anatomic counterpart, and it reflects the tendency of the muscle to develop tension before actual shortening occurs.
Developed force is inversely proportionate to velocity, and both are increased by elevations in intracellular Ca++. The maximal velocity of contraction occurs when there is no load on the system. Peak force is developed during the isometric phase of the contraction.
Contractility is defined as the change in developed force at any given fiber length, independent of preload and afterload. Contractile force is altered by changing intracellular Ca++ levels. Digitalis (a cardiac glycoside) increases Ca++ by inhibiting Na+/K+-ATPase. The resultant high intracellular Na+ acts to slow the electroneutral 2 Na+/Ca++ exchange and increase intracellular Ca++. Catecholamines increase influx of extracellular Ca++ during the action potential, and possibly through altering Ca++ transport by the SR. Tachycardia decreases the time for Ca++ to be resequestered into the SR, so free intracellular Ca++ levels rise. Any other agents that increase Ca++ will increase contractility, producing a positive inotropic effect.

 (271 votes, average: 1.96 out of 5)
(271 votes, average: 1.96 out of 5)